
Болестта, появата на която е свързана с недостатъци в генетичния клетъчен апарат, фенилкетонурия, е включена в малкото списъци на лечими наследствени заболявания. Пионер на това заболяване е лекар от Норвегия I.A. По-късно беше разкрито, че един ген, наречен фенилаланин хидроксилазен ген (дългата ръка на 12-та хромозома, съдържаща до 4, 5% от общия клетъчен ДНК материал) е отговорен за развитието и протичането на заболяването. Наследственият дефект води до частична или пълна дезактивация на чернодробния ензим фенилаланин-4-хидроксилаза.
Как действа болестта фенилкетонурия
Наследствено заболяване фенилкетонурия (PKU) води до хронично отравяне на организма с токсични вещества, образувани поради нарушен метаболизъм на аминокиселините и процеса на фенилаланиново хидроксилиране. Постоянната интоксикация причинява увреждане на централната нервна система (ЦНС), чиято проява е прогресивно намаляване на интелигентността (фенилпирувинова олигофрения).
Болестта на изсичането се проявява в прекомерното натрупване в организма на фенилаланин и продуктите от неговия неправилен метаболизъм. Други фактори в развитието на фенилкетонурия включват нарушен аминокиселинен транспорт през кръвно-мозъчната бариера, ниски нива на невротрансмитери (серотонин, хистамин, допамин). При липса на навременно лечение заболяването води до умствена изостаналост и може да причини смъртта на детето.

Механизъм за развитие на заболяванията
Причинният фактор за появата на генни нарушения е метаболитният блок, който предотвратява образуването на фенилаланин-4-хидроксилаза (ензимът, отговорен за превръщането на аминокиселината фенилаланин в тирозин). Протеиногенната аминокиселина тирозин е компонент на протеините и меланиновия пигмент, следователно е съществен елемент за функционирането на всички системи на тялото, а липсата му води до ферментация.
Последствие от потискането на образуването на метаболит, причинено от мутационно инактивиране на ензима, е активирането на спомагателни пътища на метаболизма на фенилаланин. Ароматна алфа-аминокиселина в резултат на дефектни метаболитни процеси се разпада на токсични производни, които при нормални условия не се образуват:
- Фенилпирувинова киселина (фенилпируват) е мастна ароматна алфа-кето киселина, нейното образуване води до миелинизация на невронални процеси и деменция;
- Фенил-млечна киселина - продукт, образуван по време на редукцията на фенилпирувинова киселина;
- Фенилетиламин - първоначално съединение за биологично активни предаватели на електрохимични импулси, повишава концентрацията на допамин, адреналин и норадреналин;
- orthofhenylacetate е токсично вещество, което причинява нарушаване на метаболитните процеси на подобни на мазнини съединения в мозъка.
Медицинската статистика предполага, че патологично промененият ген присъства в 2% от населението, но не се проявява. Генетичен дефект се предава на дете от родители, само ако заболяването е налице и при двамата партньори, а в 50% от случаите детето става носител на мутиралия ген, оставайки здрав. Вероятността фенилкетонурия при новородени да доведе до заболяването е 25%.
Какъв тип се наследява
Болестта на срутване е генетично заболяване, наследено по автозомно рецесивен начин. Този тип наследяване означава, че развитието на признаци на вродено заболяване ще настъпи само когато детето наследява дефектна генокопия от двамата родители, които са хетерозиготни носители на модифицирания ген.
Развитието на вродено заболяване в 99% от случаите причинява мутация на гена, отговорен за кодиране на ензим, който осигурява синтеза на фенилаланин-4-хидроксилаза (класическа фенилкетонурия). До 1% от генетичните заболявания са свързани с мутационни промени, които настъпват в други гени и причиняват дефицит на дихидроптеридин редуктаза (PKU тип II) или тетрахидробиоптерин (PKU тип III).
Фенилкетонурия при деца
Класическата форма на генетично заболяване при деца в повечето случаи се проявява в външно разпознаваеми признаци, започвайки от 3-9 месеца от живота. Новородените с дефектен ген изглеждат здрави, отличителна черта е специфичният хабитус (външен вид) на детето. Тежките симптоми се появяват 6-12 месеца след раждането.
PKU тип II се характеризира с факта, че първите клинични симптоми се появяват след 1, 5 години от момента на тяхното раждане. Симптомите на заболяването не изчезват след диагностициране на генетични аномалии и началото на диетата. Този тип вродено заболяване често води до смърт в рамките на 2-3 години от живота на детето. Най-честите симптоми на PKU тип II са:
- изразени отклонения в умственото развитие;
- хиперрефлексия;
- нарушение на двигателните функции на всички крайници;
- синдром на неконтролирани мускулни контракции.
- висока степен на умствена изостаналост;
- ясно намален размер на черепа по отношение на други части на тялото;
- мускулна спастичност (с възможна пълна неподвижност на крайниците).

Прояви на болестта на сечта
В хода на клиничните проучвания и наблюдения се предполага, че ефектите на токсичните производни на метаболизма на фенилаланин причиняват намаляване на интелектуалните способности, което е прогресивно и може да доведе до деменция (олигофрения, идиотизъм). Сред предполагаемите причини за необратими нарушения на мозъчната активност, най-разумно е липсата на невротрансмитери, които предават импулси между неврони, причинени от намаляване на нивото на тирозин.
Точната причинно-следствена връзка между наследственото заболяване и мозъчните нарушения все още не е идентифицирана, както и механизмът на развитие, дължащ се на фенилкетонурия на такива психични състояния като екопрексия, ехолалия, пристъпи на гняв и раздразнителност. Данните от резултатите от теста показват, че фенилаланинът има директен токсичен ефект върху мозъка, което също може да доведе до намаляване на интелигентността.
Физика и фенотипни особености
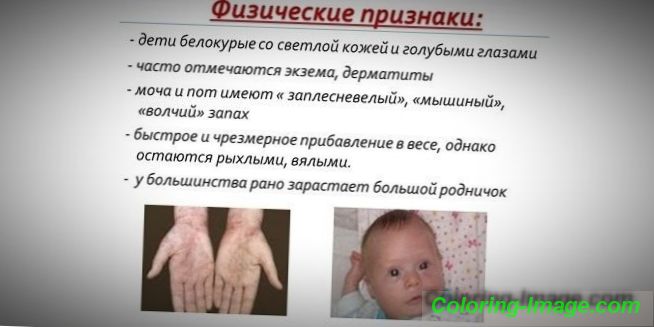
Поради факта, че пигментът на кожата и косата зависи от нивото на тирозин в хепатоцитните митохондрии, а фенилкетонурията спира конверсията на фенилаланин, пациентите с това заболяване имат фенотипни признаци (рецесивни симптоми). Повишен мускулен тонус причинява отклонение във физиката - става диспластичен. Отличителните външни признаци на фенилкетонурия включват:
- хипопигментация - светла кожа, бледосини очи, избелена коса;
- крайници, сини;
- намален размер на главата;
- специфична позиция на тялото - когато се опитвате да стоите или седнете, детето поема позита на „шивач“ (ръцете и краката са свити в ставите).
Симптоми на заболяването
С навременното откриване болестта на Felling може да се лекува успешно чрез регулиране на храненето и развитието на детето се извършва в съответствие с възрастовата му група. Трудността при откриването на генна мутация е, че е трудно да се открият ранни признаци дори за опитен педиатър. Тежестта на симптомите на вродени заболявания нараства с израстването на детето, тъй като употребата на протеинови храни допринася за развитието на разстройства на ЦНС.
Признаци при новородени
През първите дни от живота на детето е трудно да се открият признаци на патологични аномалии - бебето се държи естествено и няма забавяне в развитието. Симптомите на заболяването за първи път започват да се появяват след 2-6 месеца след раждането. Родителите трябва да бъдат предупредени за поведението на бебето, което се характеризира с ниска активност, летаргия или, обратно, тревожност, хипер възбудимост.
С началото на кърменето протеините започват да се вливат в тялото на новороденото с мляко, което служи като катализатор за появата на първите признаци, които ясно показват, че болестта е започнала да напредва. Специфичните клинични прояви на заболяването включват:
- упорито повръщане (често сбъркано с вродено свиване на пилора);
- честа регургитация;
- липса на реакция на външни стимули;
- мускулна дистония (намалено мускулно напрежение);
- конвулсивен синдром (гърчове с епилептичен или неепилептичен характер).
Симптоми при деца след 6 месеца
Ако проява на генетично заболяване не се появи (или не беше забелязана) през първите 6 месеца от момента на раждането на детето, то след този период вече е възможно да се определи точно психомоторното изоставане. Симптомите на генетични нарушения, причинени от дефицит на ензими при деца над шест месеца, са:
- намаляване на активността (до пълно безразличие);
- никакви опити да се изправят сами;
- специална миша миризма на кожата (миризмата на мухъл възниква поради екскрецията на токсични производни на фенилаланин през потните жлези и урината);
- загуба на способността на родителите за визуално разпознаване;
- лющене на кожата;
- появата на дерматит, екзема, склеродермия.
Прогресията на заболяването при липса на лечение в детска възраст
Ако не са открити аномалии в развитието в ранна детска възраст и не е проведено подходящо лечение, тогава болестта започва активно да се развива и често води до увреждане. Липсата на терапия в ранен стадий на заболяването предизвиква появата на следните симптоми на заболяването още на възраст 1, 5 години:
- микроцефалия (намален размер на мозъка);
- прогнатия (изместване на горната зъбна част напред);
- късно заздравяване;
- хипоплазия на емайла (изтъняване или пълно отсъствие на зъбен емайл);
- забавено развитие на речта до пълна липса на реч;
- 3, 4 степен на олигофрения (умствена изостаналост, умствена изостаналост);
- вродени сърдечни дефекти (дефекти в структурата на сърдечния мускул, части от сърцето, големи съдове);
- нарушения на вегетативната система (акроцианоза, прекомерно изпотяване, артериална хипотония);
- запек.
Причини и провокиращи фактори
За мутации с автозомно рецесивен модел на наследяване, дефектният ген трябва да се наследи от двамата родители. Генетичните заболявания от този тип се срещат с еднаква честота при новородените момчета и момичета. Патогенезата на PKU се определя от метаболитно нарушение на фенилаланин, което може да се прояви в 3 форми. Само класическа фенилкетонурия тип I може да се лекува с диетична терапия.
Атипичните форми на заболяването не могат да бъдат излекувани чрез регулиране на диетата. Тези отклонения са причинени от дефицит на тетрахидроптерин, дехидроптерин редуктаза (по-рядко пирувоил тетрахидроптерин синтаза, гуанозин-5-трифосфат циклохидролаза и др.). Повечето случаи на летални резултати са регистрирани при пациенти с редки вариации на ФКУ, докато клиничните прояви на всички форми на заболяването са сходни. Рискът от раждане на дете с мутиран ген на фенилаланин хидроксилаза се увеличава, ако родителите му са близки роднини (в близки бракове).
диагностика
Ако се подозира генетично заболяване, се установява диагноза въз основа на комбинация от данни, получени от проучване на историята на заболяването - генеалогична информация, резултати от клинични и медицински генетични изследвания. За навременното откриване на вродени заболявания (PKU, кистозна фиброза, галактоземия и др.) Е разработена задължителна програма за масов скрининг за всички новородени (неонатален скрининг).
Ако бъдещите родители знаят за превоза на мутиралия ген, съвременната медицина предлага начини за откриване на дефект по време на бременност (пренатална диагностика на плода чрез инвазивен метод). За отделянето на фенилкетонурия от видове според тежестта се използва условна класификация, която се основава на нивото на фенилаланин в течност без фибрин, получена от кръвната плазма:
- Тежка фенилкетонурия - 1200 µmol / l.
- Средната стойност е 60-1200 μmol / l.
- Светлина (не изисква лечение) - 480 µmol / l.
Скринингов тест
Идентифицирането на генетични аномалии се наблюдава в няколко етапа. На първия етап в родилния дом, за всички бебета на 3-5 дни от живота, периферната кръв се взима (от петата) за изследване. Материалът се полага върху хартиен носител и се изпраща в биохимичната лаборатория, където се извършва биохимичен анализ. На втория етап на скрининговия тест се установява, че концентрацията на фенилаланин е нормална.
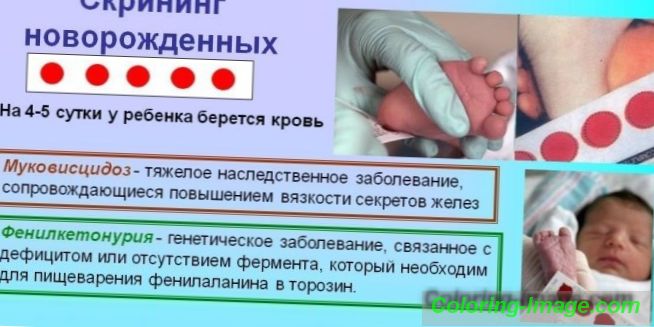
Ако не бъдат открити патологични промени, диагнозата завършва и се вписва в картата на детето. При наличие на отклонения от нормата, резултатите от диагнозата се изпращат на педиатър, за да се направи изясняващ преглед на кръвната проба на новороденото. Здравето на бебето зависи от навременното и точно изпълнение на всички мерки за идентифициране на отклонения. Ако диагнозата бъде потвърдена след повторно изследване, родителите на детето ще бъдат насочени към клиниката за педиатрична генетика за целите на лечението.
Анализи и проучвания за потвърждаване на диагнозата
Повтарящата се диагностика, когато се открият аномалии по време на първоначалния скринингов тест, се извършва чрез многократни тестове. В допълнение към определянето на съдържанието на фенилаланин в кръвта, методите за диагностициране на ФКУ при деца и възрастни включват:
- Тест за изсичане - определяне на фенилпирувинова киселина в урината чрез добавяне на ферихлорид към биоматериала (настъпва синьо-зелено оцветяване);
- Guthrie test - оценка на степента на реакция на микроорганизмите към метаболитни продукти или ензими, съдържащи се в кръвта на пациента;
- хроматография - изследване на химичните свойства на веществата, разпределени между две фази;
- флуориметрия - излъчване на биоматериал с монохроматично лъчение за определяне на концентрацията на съдържащите се в него вещества;
- електроенцефалография - диагностика на електрическата активност на мозъка;
- магнитен резонанс е възбуждането на атомните ядра на клетките чрез електромагнитни вълни и измерването на техния отговор.
Лечение на класическа фенилкетонурия
Основата на терапията с фенилкетонурия е ограничаването на консумацията на продукти, които са източник на животински и растителни протеини. Единственият метод за успешно лечение е диетична терапия, адекватността на която се оценява от съдържанието на фенилаланин в кръвния серум. Максимално допустимото ниво на аминокиселини при пациенти от различни възрастови групи е:
- при новородени и деца до 3 години - до 242 µmol / l;
- при деца в предучилищна възраст - до 360 µmol / l;
- при пациенти на възраст от 7 до 14 години - до 480 µmol / l;
- при юноши до 600 μmol / l.
Ефективността на диетата зависи от етапа на коригиране на заболяването. При ранна диагностика на вродена патология се предписва диетична терапия от 8-та седмица от живота (след този период започват необратими промени). Липсата на своевременни мерки води до усложнения и намаляване на нивото на интелигентност с 4 пункта за 1 месец от момента на раждането до началото на лечението.

Поради факта, че терапевтичната диета за фенилкетонурия предполага пълно елиминиране на животинските протеини от диетата, е необходимо да се използват други източници на есенциални аминокиселини, както и витамините от група В, съдържащи калций и фосфор минерални съединения. Продуктите, обозначени като добавки към диета без протеини, включват:
- протеинови хидролизати (Amigen, Aminazole, Fibrinosol);
- свободна от фенилаланин смес, наситена с незаменими аминокиселини - тетрафен, без фенил.
Заедно с оздравителните мерки за отстраняване на причината за нарушеното функциониране на организма, трябва да се извърши симптоматично лечение, насочено към елиминиране на дефекти в речта и нормализиране на координацията на движенията. Комбинираната терапия включва физиотерапевтични процедури, масаж, помощ от логопед, психолог, гимнастически упражнения. В някои случаи, заедно с диетичната терапия, се посочва употребата на антиконвулсанти, ноотропни и съдови лекарства.
Особености на лечението на атипични форми
Фенилкетонурия тип II и тип III не може да се лекува с диета с ниско съдържание на протеини - нивото на фенилаланин в кръвта остава непроменено, когато приемът на протеин в организма е ограничен или клиничните симптоми напредват дори и с намаляване на нивото на аминокиселините. Ефективна терапия на тези форми на заболяването се извършва с помощта на:
- тетрахидробиоптерин - фактор на засегнатия ензим;
- синтетични аналози на тетрахидробиоптерин - тези вещества проникват по-добре в кръвно-мозъчната бариера;
- лекарства за заместителна терапия - не елиминират причината за фенилкетонурия, но поддържат нормалното функциониране на организма (леводопа заедно с карбидофа, 5-хидрокситриптофан, 5-формил тетрахидрофолат);
- хепатопротектори - подпомагат функционирането на черния дроб;
- антиконвулсанти;
- въвеждането на гена фенилаланин хидроксилаза в черния дроб - експериментален метод.
Особености на храненето на новородените и диетична терапия
През първата година от живота на детето с ФКУ кърменето е приемливо, но неговото количество трябва да бъде ограничено. До 6 месеца приемливото ниво на прием на фенилаланин е 60-90 мг на 1 кг тегло на бебето (100 г мляко съдържа 5, 6 мг фенилаланин). От 3 месеца диетата на детето трябва постепенно да се разширява, за да включва плодови сокове и картофено пюре.
Деца от 6 месеца позволяват въвеждането в диетата на растително пюре, овесена каша (от саго), безцелулозни целувки. След 7 месеца можете да дадете на бебето ниско протеинова паста, от 8 месеца - хляб, който не съдържа протеини. Не е установена възрастта, до която е необходимо да се ограничи приема на протеини в тялото на болно дете. Докторите досега обсъждат осъществимостта на терапията с диета през целия живот, но се съгласяват, че е необходимо да се придържате към диетична диета поне 18 години.
Фенилкетонурия, диагностицирана при жена, не е причина да се откаже да роди дете. Бременните майки с ФКУ за предотвратяване на увреждане на плода по време на бременност и предотвратяването на възможни усложнения са необходими преди планираното зачеване и при пренасяне на дете да следва диета с ограничение на фенилаланин (нивото му в кръвта трябва да бъде до 242 µmol / l).
Без лактоза формула за бебета
Диета с фенилкетонурия се основава на значително намаляване на дозата на естествения протеин в ежедневната диета, но тялото на новороденото бебе не може да се развие нормално при липса на необходимите микроелементи. За да се отговори на нуждите на бебето в протеини, се използват аминокиселинни смеси без лактоза, които според руското законодателство трябва да се предоставят безплатно на пациентите.
Толерантността на бебетата към фенилаланин през първата година от живота се променя бързо, така че е необходимо да се следи концентрацията му в кръвта на детето и да се правят корекции в диетата. Смесите са предназначени за специфични възрастови групи:
- за бебета до една година се назначават Афинелак 15, Аналог-СП, ФКУ-1, ФКУ-микс, ФКУ Анамикс;
- деца над 1 година са предписани смеси, обогатени с витамини и минерали с високо съдържание на протеини - PKU Prima, P-AM Universal, PKU-1, PKU-2, XP Maxameyd, XP Maxamum.

Диетични продукти за попълване на протеиновите резерви
Един от основните компоненти на хранителната диета с фенилкетонурия е храни с ниско съдържание на протеини на базата на нишесте. Тези добавки съдържат казеинов хидролизат, триптофан, тирозин, метионин, азот и осигуряват дневните нужди на детето, което е необходимо за нормалното развитие и растеж. Специализирани продукти, които компенсират липсата на основни минерали и аминокиселини, когато те липсват в диетата, са:
- Berlofen;
- Tsimorgan;
- Minafen;
- Апонте.
Диета за деца в предучилищна възраст и ученици
Тъй като тялото се адаптира към фенилаланин, децата на възраст над 5 години могат постепенно да намалят диетичните ограничения. Разширяването на диетата се осъществява чрез въвеждане на зърнени храни, млечни продукти, месни продукти. Учениците в гимназията вече имат висока толерантност към фенилаланин, така че в тази възраст можете да продължите да разширявате диетата, докато е необходимо да следите отговора на всички промени в диетата. За да контролирате състоянието на детето, се използват следните методи:
- оценка на неврологични показатели, психологическо състояние;
- мониторинг на работата на електроенцефалограмите;
- определяне на нивото на фенилаланин.
Групи продукти за PKU
Диетата на пациенти с ФКУ, заедно с ниско протеинови скорбялни храни и терапевтични смеси, също включва продукти от естествен произход. При съставянето на менюто трябва ясно да се изчисли количеството консумиран протеин и да не надвишава препоръчваната от лекаря доза. За да се изключат токсичните ефекти върху организма, са разработени 3 списъка с продукти, които съдържат забранени (червени), не препоръчвани (оранжеви) и разрешени (зелени) позиции.
Червен списък
Фенилкетонурия се развива на фона на отсъствието на ензим, който превръща фенилаланин в тирозин, така че високото съдържание на протеин е причината за приписване на продукти на забранения (червен) списък. Позициите от този списък трябва изцяло да изключват диетата на пациента, PKU:
- месо;
- вътрешни органи на животни, карантии;
- колбаси, колбаси;
- морски дарове (включително риба);
- яйца от всички птици;
- ферментирали млечни продукти;
- ядки;
- плодове от бобови и зърнени култури;
- соеви продукти;
- Съдове, съдържащи желатин;
- сладкарски изделия;
- аспартам.
Оранжев списък
Продукти, които трябва да се дозират в тялото на дете, диагностицирано с ФКУ, са включени в оранжевия списък. Включването в хранителния режим на позиции от този списък е приемливо, но в строго ограничени количества. Въпреки че тези продукти не съдържат много протеини, мога да повиша и нивото на фенилаланин, така че тяхното използване не се препоръчва:
- консервирани зеленчуци;
- ястия от картофи и ориз;
- зеле;
- мляко;
- шербет.
Зелен списък
Продуктите, които не съдържат протеини, са разрешени за употреба от пациенти с диагноза фенилкетонурия без ограничения. Преди да закупите продукти от зеления списък, трябва да разгледате състава, показан на опаковката, и да се уверите, че няма аспартамова боя, съдържаща фенилаланин:
- плодове;
- зеленчуци (с изключение на картофи и зеле);
- плодове;
- зелени;
- нишестени зърнени храни (саго);
- мед, захар, конфитюр;
- Брашно от брашно от царевица или ориз;
- масла, мазнини (кремави, зеленчукови, маслинови).
Как да контролирате нивата на фенилаланин в кръвта
Фенилкетонурията е неизлечима болест, която може да се превърне във фаза на стагнация чрез използване на диетична терапия и терапевтични мерки. При изменении условий жизни, нарушении режима питания болезнь может опять обостриться, поэтому больным требуется пожизненное наблюдение. Процесс контроля заключается в периодическом определении уровня фенилаланина в крови. Частота сдачи анализов зависит от возраста пациента:
- до 3 месяцев – скрининг крови надо делать еженедельно до получения устойчивых результатов;
- от 3 месяцев до 1 года – 1-2 раза в месяц;
- от 1 до 3 лет – 1 раз за 2 месяца;
- старше 3 лет – ежеквартально.

Кровь для анализов сдается через 3-4 часа после приема пищи. Помимо скрининга развитие ФКУ контролируется путем определения нутритивного статуса, физического, эмоционального развития больного, уровня интеллектуальных способностей и развитости речи. По результатам наблюдений может возникнуть необходимость дополнительной диагностики с привлечением соответствующих специалистов.